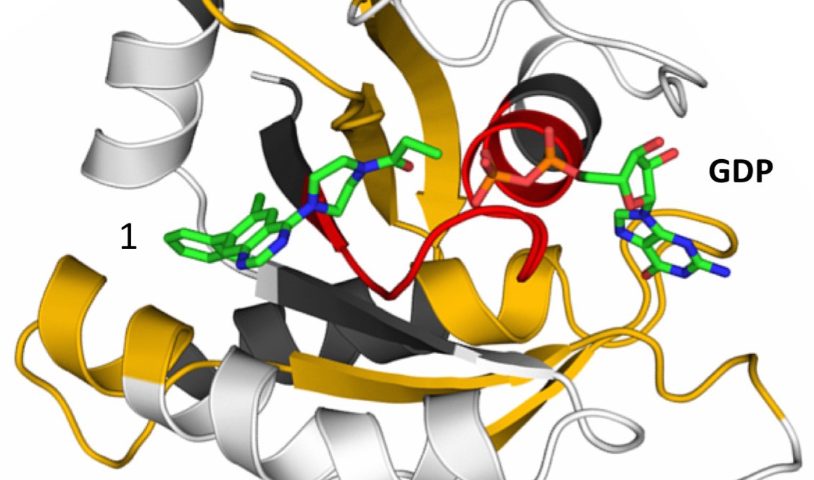
The crystal structure of mutant KRAS G12C in complex with a KRAS inhibitor of the quinazoline chemotype and GDP.
While genomic and functional characterization of cancer continues to lead to the discovery of new potential targets, advances in cancer treatment are limited by the number of available drugs that can effectively target them. Even so, the range of targets that are considered “druggable” is continuously evolving.
Promising Breakthroughs
The Gray Lab has had success in developing compounds to antagonize targets that were previously considered intractable. One such breakthrough was the development of selective covalent CDK7 inhibitors. As CDK7 is implicated in transcriptional regulation, selective inhibition targets the transcriptional dependencies of several cancers, for example, neuroblastoma, small cell lung cancer, and triple negative breast cancer. The first-in-class CDK7 inhibitor THZ1, developed in the Gray Lab, has shown promise in preclinical models. More recently, Syros Pharmaceuticals has developed clinical grade CDK7 inhibitors which have led to clinical trials. We are currently testing CDK7 inhibitors in both genetically engineered mouse models (GEMMs) and in patient-derived xenografts (PDXs) of pancreatic cancer.
We also have ongoing efforts to target KRAS directly. We have been successful in targeting the specific missense mutant KRAS G12C with a tool compound. While G12C mutation is a rare event in pancreatic cancer (<2% of cases), the development of an effective mutant-KRAS specific inhibitor suggests similar compounds might be discoverable for more common KRAS alterations, e.g., KRAS G12D/V/R, which are identified in >90% of KRAS-mutant pancreatic ductal adenocarcinomas (PDAC).
Screening Experimental Compounds
In addition to specific candidate compounds such as those directed toward CDK7, the Hale Family Center For Pancreatic Cancer Research is using libraries of experimental compounds from the Gray Lab for systematic screening in organoid models.
If a target is identified by genetic means in loss-of-function screens and validated, but no suitable inhibitor exists, the Gray Lab will attempt to generate a novel inhibitory compound using structure modeling.
In collaboration with Dr. Jay Bradner, MD (Novartis), we have also developed a generalizable strategy for chemical degradation. These selective degraders act as bifunctional small molecules: one end of the molecule recruits E3 ubiquitin ligases, e.g., cereblon, via a phthalimide moiety, while the opposite end of the molecule allosterically binds a target protein. This is an especially exciting development as it is our hope that this technology could be used to directly target and degrade mutant KRAS.
