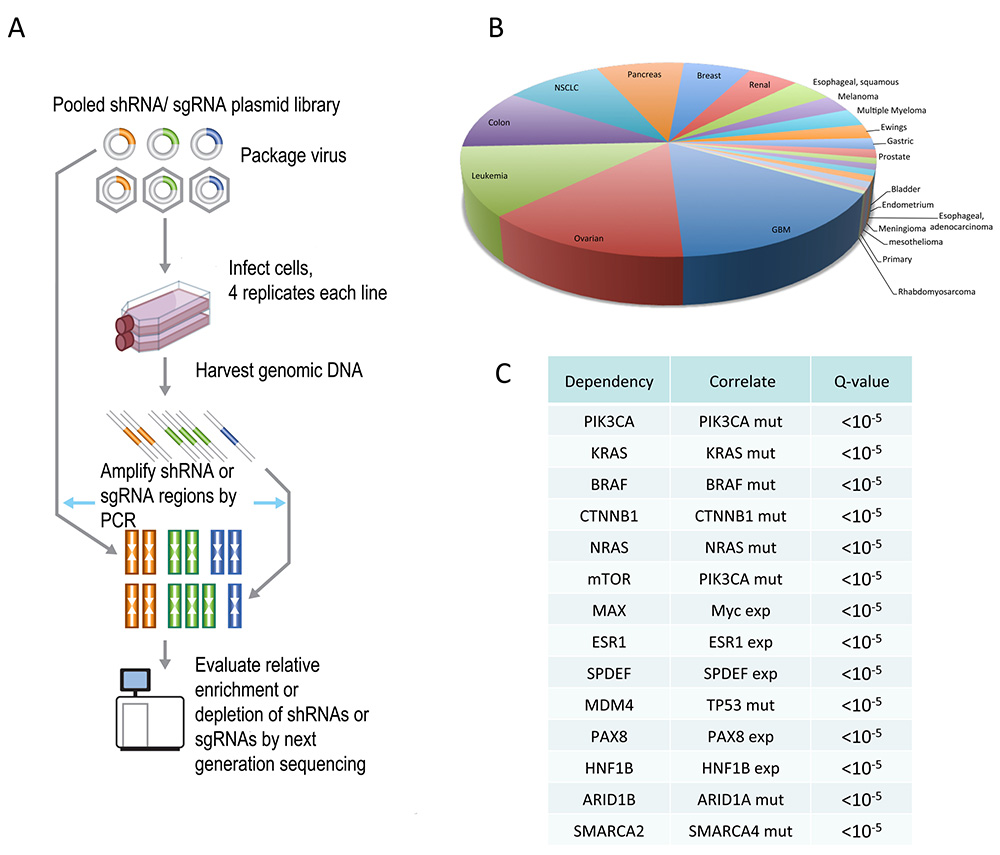
Project Achilles A. Schematic of the Project Achilles screening pipeline based on RNAi or CRISPR-Cas9 B. Lineages represented in the current Project Achilles dataset. C. Dependencies of known and validated genetic and therapeutic paradigms.
Development of pancreatic ductal adenocarcinoma (PDAC) is a multistep process that commonly involves mutational activation of KRAS, sequentially followed by loss of the CDKN2A, TP53, and SMAD4 tumor suppressor genes. KRAS represents a key signaling switch in cancer cells. Mutation results in locking of the KRAS protein into a constitutively active conformation which leads to uncontrolled downstream signaling. Even after transformation, cancer cells continue to require KRAS for survival and proliferation.
Identifying A Target
While historical attempts at targeting KRAS directly have failed, there is new hope in the field of KRAS targeting (e.g., KRAS G12C inhibitors). Alternative approaches are focused on blocking downstream signaling pathways, e.g., interrupting MAPK signaling with either MEK or ERK inhibitors.
While MEK inhibitors have elicited partial responses in some cancers, e.g., in melanoma, they have proven ineffective in PDAC. We have performed large-scale screens to identify modifiers of MEK inhibitor sensitivity hoping to find clues as to effective combination therapies that could overcome MEK inhibitor resistance. In preclinical models of lung cancer, we have shown that the combination of a MEK inhibitor (MEKi) and a TBK1 inhibitor (TBK1i) is an effective combination. We are exploring if the MEKi/TBK1i combination might also be effective in the context of PDAC.
We are also interrogating novel PDAC cancer cell vulnerabilities by performing RNAi or CRISPR-Cas9 based loss-of-function screens in cell lines and tumor organoids. Targets identified in vitro are first confirmed in multiple cell lines and subsequently validated in vivo using genetically-engineered mouse models (GEMMs) or patient-derived xenografts (PDXs).
